
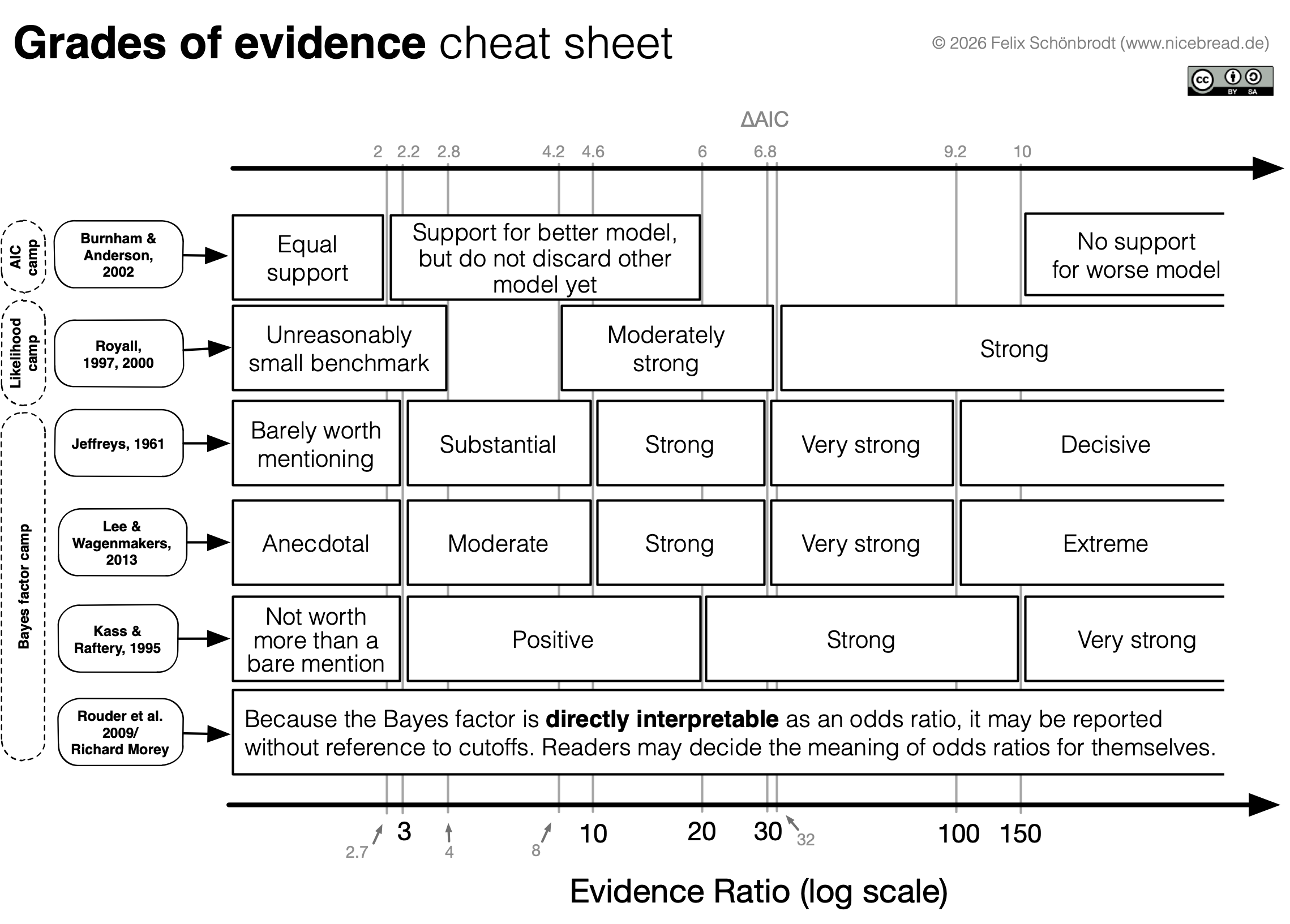
Grades of Evidence Cheat Sheet
Jun 11, 2026
Professor at the Department of Psychology, Ludwig-Maximilians-Universität München, Germany. Managing director of the LMU Open Science Center and board member of the META-REP priority program (“A meta-scientific research program to analyse and optimise replicability in the behavioral, social, and cognitive Sciences”). Interested in Open Science, Metascience, implicit motives, machine learning, responsible research assessment (CoARA), solarpunk, piano, and cooking.
![]()
I embrace the values of openness and transparency in science. I believe that such research practices increase the informational value and impact of our research, as the data can be reanalyzed and synthesized in future studies. Furthermore, they increase the credibility of the results, as an independent verification and replication is possible.
For this reason, I developed and signed a Commitment to Research Transparency and Open Science.
You can sign, too!
